
Aaron Dines
The hydration barrier is one of the principle quantities governing the translocation of ions through nanoporous materials, it results from the reorganisation of water in the electric field of a charge in solution. Typically, a description is achieved through brute-force molecular dynamics simulations in the literature, but at the cost of computational efficiency and explanatory value; molecules are simulated individually with a computer programme. Recently, an analytical method has been proposed that allows for a qualitative description of water distributions at far less computational cost. We use this method to investigate the hydration patterns for an ion moving through a hexagonal-boron nitride nanopore, and investigate the effects of pore geometry, skew, twist, size and bend of the lattice. This particular lattice is of interest due to its uneven charge distribution, resulting in highly complex hydration patterns. We then use the hydration patterns to calculate the free energy profile, which characterises the kinetics of translocation and controls permeation through the nanopore. Our methods may be used to essentially design nanopores with on-demand selectivity properties, which has applications in water desalination, so-called ‘blue-energy’ harvesting, DNA sequencing and drug design.

Aaron Dines
Introduction Many modern techniques for DNA sequencing, "blue energy" harvesting and water desalination [1,2,5,6] require on-demand selective control of artificial nanopore properties. The quantities of interest being hydration patterns of water around the pore and the dehydration barrier resulting from the reorganisation of solvent around the pore and incoming ions. The main barrier to progress in this regard has been computational efficiency; typically the methods employed are through brute force molecular dynamics (MD) simulations. While capable of producing the correct behaviour, sampling different conformations is an arduous and costly process. Generally, analytical theories are far more computationally efficient, and so the methods employed involve finding a suitable analytic characterisation [4].
The system being described is governed by nonlinear equations. When an atom is added to solution the density pattern of water changes and the way in which that particular ion changes the water pattern depends on the presence of all others. This means that under normal circumstances the equations cannot be ‘decoupled’ and in order to describe the change made to one atom, the whole system needs to be simulated again. However, it is common for nonlinear equations to exhibit quasi-linear behaviour in certain circumstances. A prime example is the reduction of the nonlinear Einstein field equations to a linear Newtonian theory of gravity at low speeds and weak fields. The latter is more than capable of describing the orbit of most planets in our solar system. For this reason the first thing any physicist would try is to assume the equations behave linearly. Click here for a video about nonlinearity.
Click here for a technical discussion about the analytic approach.
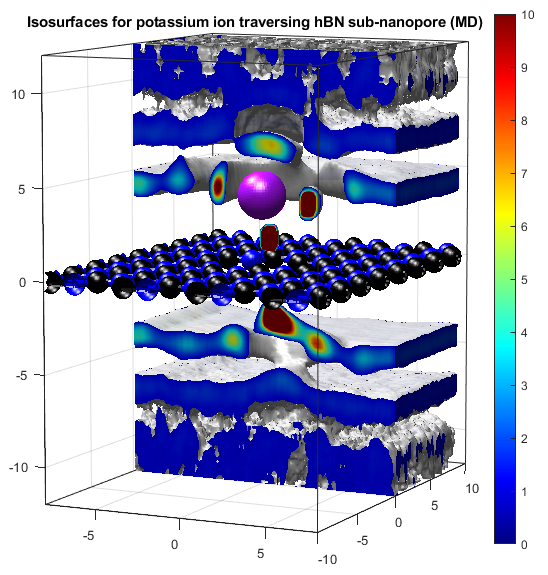
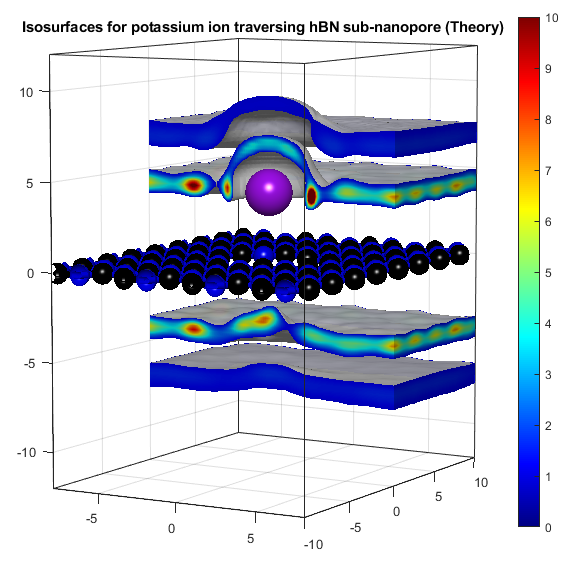
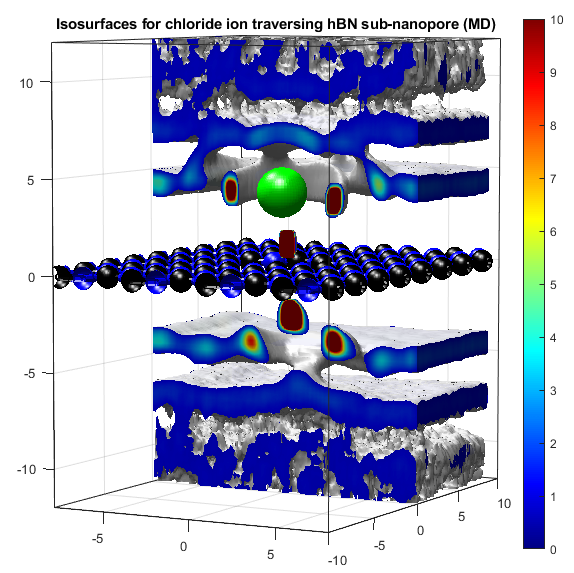
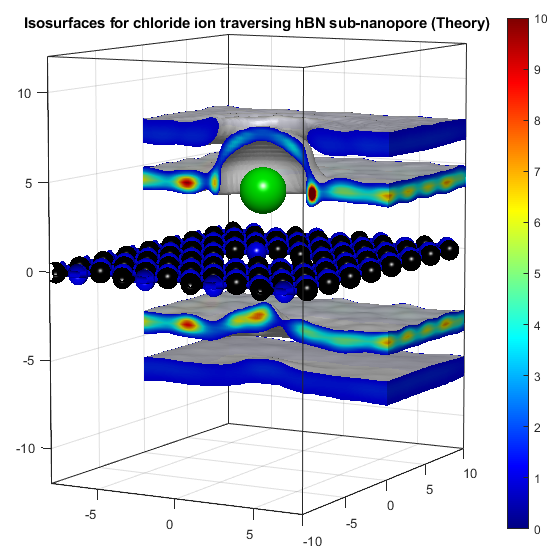
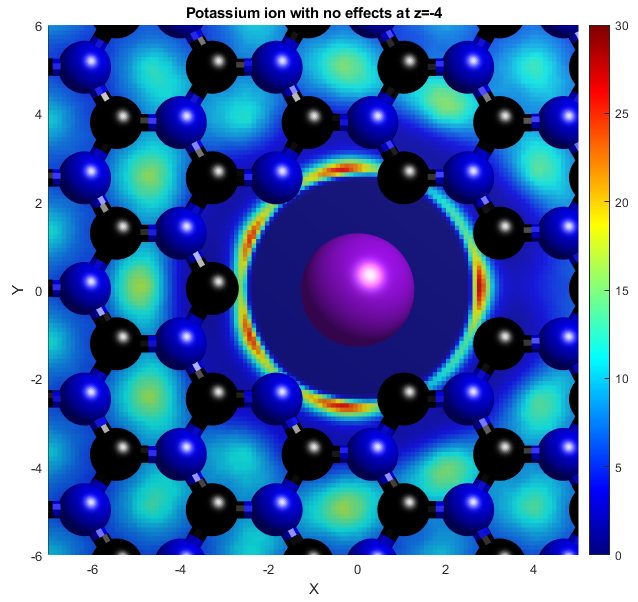
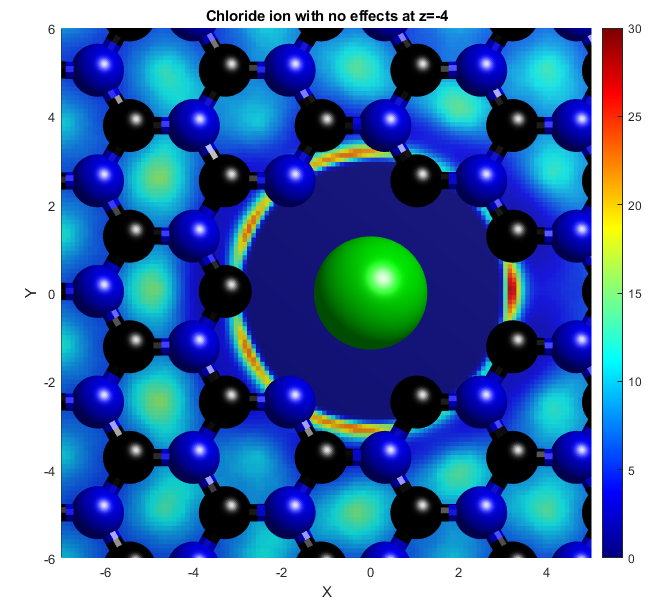

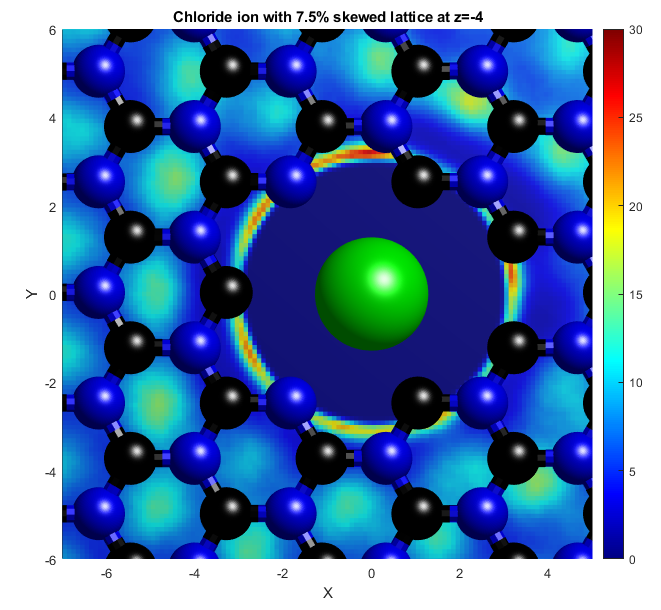
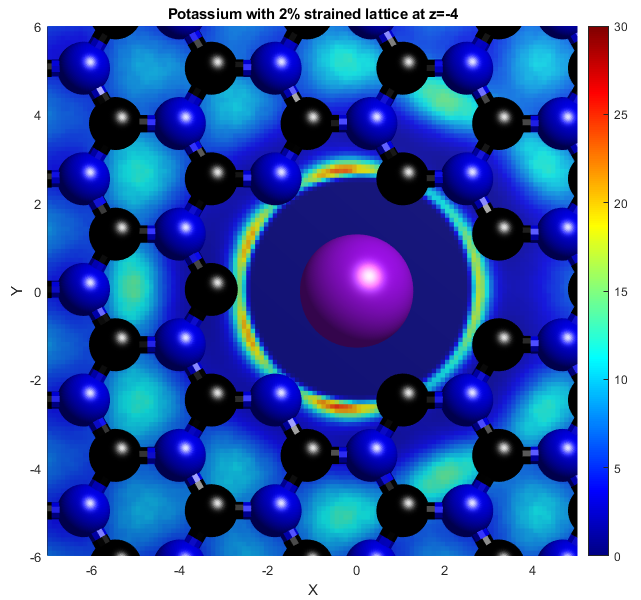
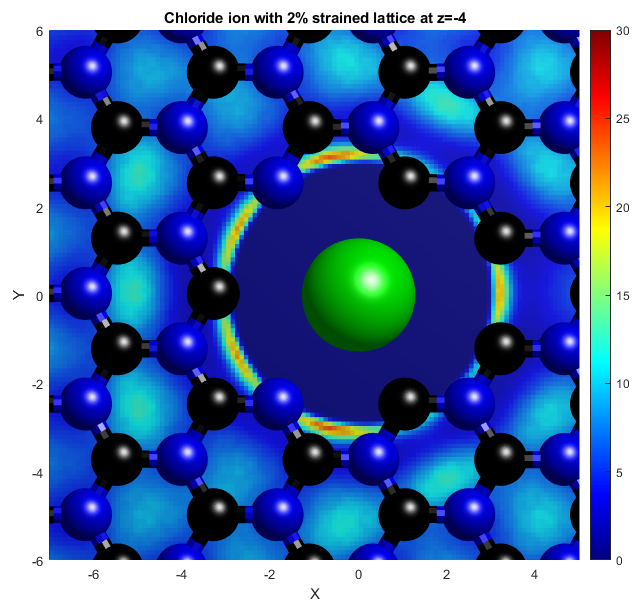
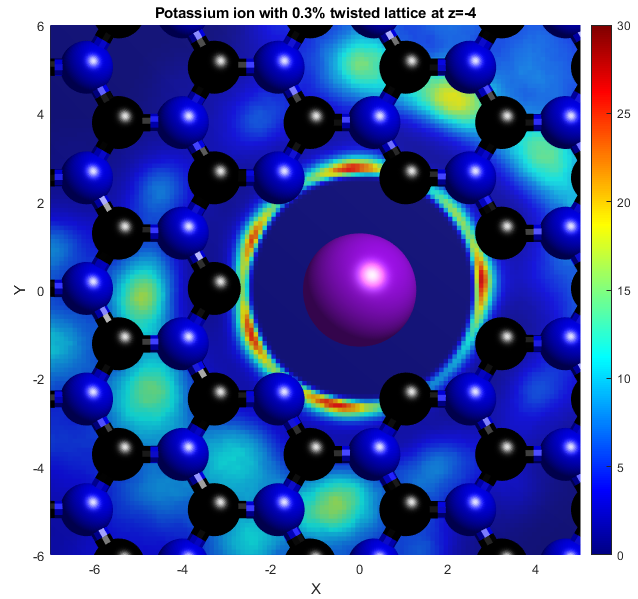
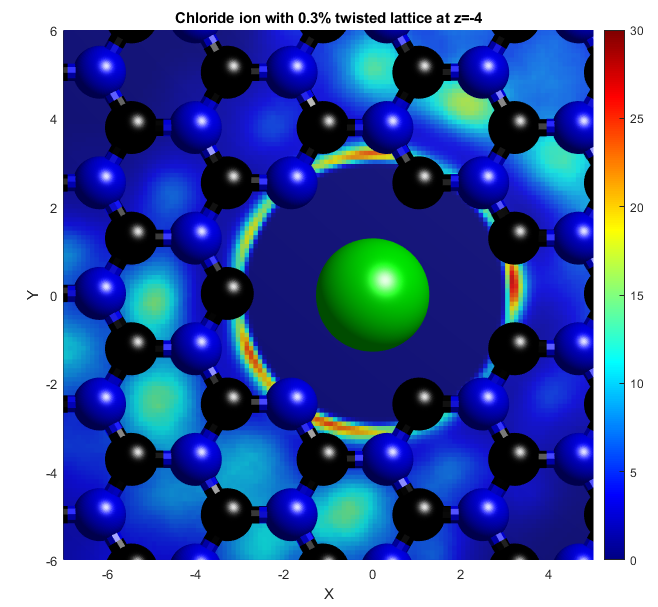
In the above, the potassium and chloride ions are represented by purple and green balls respectively. The colourbars show the density as it compares to bulk with a value of 1. The top two graphs show "isosurfaces", where the density is equal to a predefined value. Strain has the effect of expanding the lattice in all directions, increasing







